Genética de la fenilcetonuria e hiperfenilalaninemia
¿Cómo se hereda la fenilcetonuria?
La fenilcetonuria es una condición genética heredada de los dos padres.
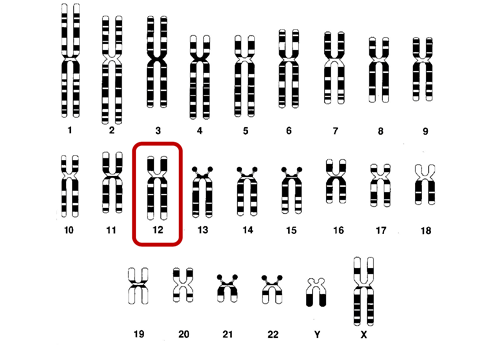
Cada célula de nuestro organismo tiene un conjunto de pequeñas estructuras llamadas cromosomas los cuales contienen miles de genes responsables del desarrollo físico y mental del individuo. Estos genes están compuestos de ADN. El óvulo materno contiene 23 cromosomas y el espermatozoide paterno otros 23 cromosomas. Recibimos así 23 pares (46 en total) de cromosomas que contienen los genes procedentes la mitad del padre y la otra mitad de la madre. Por lo tanto, todas las células de nuestro organismo tienen los 23 pares de cromosomas lo que supone entre 50.000 y 100.000 pares de genes.
¿Dónde está localizado el gen PAH?
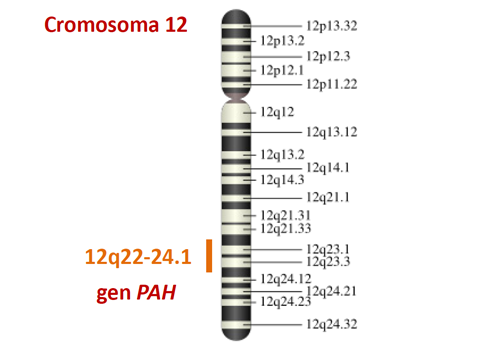
En uno de estos cromosomas (cromosoma 12) hay un gen llamadoPAH que cuando está alterado (mutado) en ambos cromosomas es responsable de la fenilcetonuria o de la hiperfenilalaninemia.
¿Por qué se producen las mutaciones?
Se estima que cada uno de nosotros porta al menos 10 genes que han sido seriamente alterados (mutados) como resultado de algún "accidente" celular. En la mayoría de los casos el resultado de estas alteraciones no se observa porque tenemos todavía el otro cromosoma que lleva una copia del gen sin alterar.
¿Qué pasa si hay un solo cromosoma mutado?
Los individuos que reciben de uno de los padres un gen alterado y el otro normal se denominan "portadores". En cambio cuando ambos padres son portadores y el hijo recibe los dos cromosomas con el gen alterado, este niño manifestará la enfermedad. Así pues, un doble portador presenta la enfermedad.
¿Qué significa herencia autosómica recesiva?
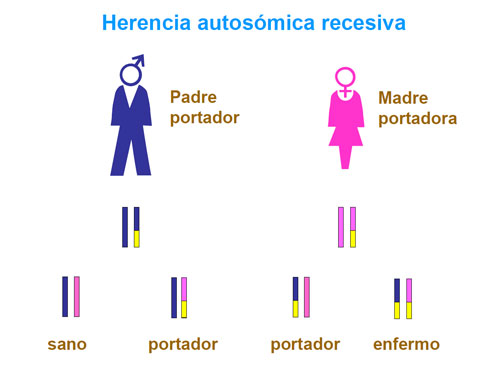
Cada vez que dos portadores conciban un hijo, la probabilidad de que éste reciba los dos cromosomas con el gen alterado es de un 25%. La probabilidad de que el niño sea sano pero portador es del 50% y, finalmente, la probabilidad de que sea sano y no portador es del 25%. Este tipo de herencia se denomina herencia autosómica recesiva y es la que presenta la fenilcetonuria.
¿Cuál es la incidencia de la PKU?
La fenilcetonuria tiene una incidencia general de 1 cada 10.000 recién nacidos vivos aunque hay variación según la zona geográfica. La frecuencia de portadores es del 2% en la población general.
¿Se pueden estudiar las mutaciones de la PKU?
Actualmente se pueden estudiar las mutaciones del gen PAH, aunque debido a lagran cantidad de mutaciones (alteraciones) distintas existentes (más de 500 descritas) en algunos casos puede resultar algo difícil y lento. Es necesario estudiar el DNA del individuo enfermo y el de sus padres. Combinando diferentes técnicas de biología molecular podemos llegar a conocer la alteración genética en el 95% de los casos.
¿Quién debe estudiarse para la PKU?
- Los niños afectos.
- Sus padres (importantes para confirmar las mutaciones del niño afecto).
- Sus hermanos sanos o afectos (posibles portadores), aún cuando puede ser más conveniente que ellos mismos lo pidan cuando tengan autonomía propia.
- Cualquier familiar a riesgo según la historia familiar.
¿Qué utilidad tienen estos estudios para las familias PKU?
- Según la mutación o mutaciones que tenga, el paciente presentará una formamás leve o más severa de la enfermedad, lo que hace que alteraciones en el mismo gen causen en ocasiones PKU (fenilcetonuria) y en otras mHPA (hiperfenilalaninemia moderada). El conocer qué mutaciones tienen nuestros pacientes nos puede ayudar a relacionar el genotipo, es decir, las mutaciones en el gen PAH, con el fenotipo, es decir, con las manifestaciones clínicas. Esto nos ayudará a predecir la gravedad de la enfermedad.
- Por otro lado, el conocimiento de las mutaciones nos permitirá seleccionar a los pacientes candidatos a tratamiento con BH4 [ ver Tratamiento ], el cofactor de la enzima fenilalanina hidroxilasa, pues los pacientes con mutaciones leves que permiten cierta actividad residual de la enzima PAH son los que responden mejor a esta terapia.
- Una vez conocidas las mutaciones en la familia PKU o HPA y tras realizar el estudio familiar podremos ofrecer consejo genético (el riesgo para la descendencia) tanto al paciente como a sus familiares (padres, hermanos, etc.).
- El hecho de conocer las mutaciones resulta imprescindible para poder ofrecer un diagnóstico prenatal, que en la PKU no está justificado dado el buen pronóstico de la enfermedad diagnosticada y tratada precozmente.
¿Cómo se realiza el diagnóstico prenatal?

Ante el embarazo de una pareja en que ambos sean portadores y se conozca la alteración en ambos casos se puede realizar el estudio de mutaciones envellosidad corial (semana 8-11 de embarazo) o en líquido amniótico(semana 16 de embarazo) y conocer si se trata de un feto sano, portador o afecto.


